SC22 Gordon Bell Finalist:DGDFT软件实现250万原子超大规模复杂金属异质结体系的电子结构计算模拟
当地时间11月15日下午,瀚海量子首席科学家胡伟凭借DGDFT高性能计算应用成果强势入围国际戈登•贝尔奖提名,并参加了在美国得克萨斯州达拉斯市召开的全球超级计算大会(SC22)的线上答辩,报告了在新一代神威超级计算机上采用低标度高精度第一性原理计算软件DGDFT首次实现250万原子复杂金属异质结(锂钠金属合金)的超大规模材料模拟。戈登•贝尔奖是国际上高性能计算应用领域的最高学术奖项,被誉为“超算领域的诺贝尔奖”,由ACM(美国计算机学会)每年评选和颁发,奖励在高性能计算应用领域具有突出成就的工作,具有巨大的国际影响力。此项成果由合肥微尺度物质科学国家研究中心杨金龙院士课题组胡伟团队与计算机科学与技术学院安虹教授课题组联合攻关,在崂山实验室(原青岛海洋科学与技术试点国家实验室)、中国科学院计算技术研究所、北京大学、中国科学院软件研究所、齐鲁工业大学,以及国家并行计算机工程技术研究中心相关研究人员的紧密配合下完成。

图1 获ACM Gordon Bell奖提名
先进材料是国民经济的基石,是实现制造业转型升级的重要基础。由于新型材料的研发难度较大,用于验证材料性质的物理实验设计复杂、代价昂贵。通过输入材料的结构信息,利于量子力学的第一性原理计算就可以较为准确地预测已知材料的基态结构和基本的物理化学性质,并实现原子级别的精准控制,是二十一世纪解决实验理论问题和预测新材料结构性能的最具竞争力的方法和技术途径。该研究方法不需要开展真实的实验,不仅可以极大地节省实验成本,缩短新材料的开发周期,而且可以为材料的制备和改性、新材料的开发,以及对极端环境下材料性质的研究提供有效的理论指导。但是,由于第一性原理材料模拟的计算复杂度会随着模拟材料尺度而急剧增加,研究人员对软件的性能和计算资源的需求越来越大。超级计算机和高性能计算技术的快速发展为第一性原理计算的发展提供了机遇,使其正在凝聚态物理、材料科学、化学和生物等研究领域发挥着越来越重要的作用。
采用基组离散Kohn-Sham方程是实现第一性原理电子结构计算的基础。传统基于平面波基组的第一性原理材料模拟具有三阶的计算复杂度,很难通过大规模并行加速应用于复杂的大体系。基于局域原子轨道基组的线性标度算法可以实现大尺度模拟,但是往往精度不够并难以应用于金属体系,同时还面临非规则稀疏矩阵难以实现高度并行计算的难题。全新的自适应性局域基组(adaptive local basis, ALB),在实空间严格截断且正交,结合了平面波(正交完备性)和数值原子轨道(局域性)各自的优点,既有媲美平面波的高精度又能利用适合于大尺度计算的线性标度算法。并且,由ALB基组构造的哈密顿矩阵具有固定的三对角块状的稀疏特性,适合于实现高度并行。中国科大与中科院计算所、中科院软件所等单位合作,以ALB基组和间断伽辽金有限元方法为基础,发展了第一性原理计算软件DGDFT(discontinuous Galerkin density functional theory),结合多级并行优化设计和最先进的稀疏矩阵求解器PEXSI的低标度对角化算法,在数值算法、稀疏矩阵并行和数据通讯等方面进行一系列关键技术创新,采用在新一代神威超级计算机上自主设计的DGDFT软件首次实现了250万原子复杂金属体系的高精度第一性原理电子结构模拟,达到了介观尺度(> 100 nm)范围,可用于设计基于二维材料的新一代电子晶体管。
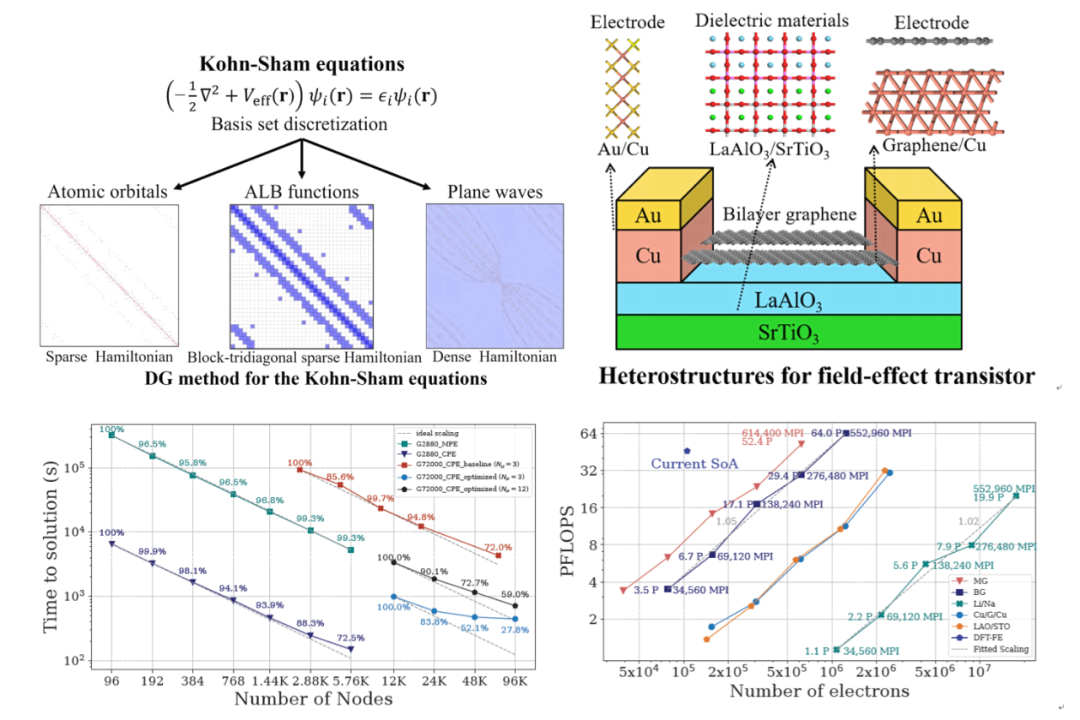
DGDFT软件的先进性体现在:(1)相比于国际上同类第一性原理计算软件,DGDFT软件具有低标度、高精度以及高并行可扩展性优势,克服了常规DFT方法难以应用于复杂金属体系、稀疏矩阵并行扩展性差等多个一直困扰材料模拟领域和高性能计算领域的难题。(2)尽管间断伽辽金方法广泛应用于求解偏微分方程,但是目前只有DGDFT软件采用了该方法来求解Kohn-Sham方程。(3)通过ALB基组离散的矩阵具有三角块稀疏的固定格式,相关的研究目前仍然非常前沿,国际上尚无同类的第一性原理软件。(4)低标度PEXSI算法适合于金属体系,其对于准二维体系具有1.5次的计算复杂度。经过团队优化后的PEXSI算法可以在新一代神威超算上实现64 PFLOPS和5%的峰值,远超目前最先的HPCG稀疏矩阵性能基准测试记录(Fugaku, Summit 和新神威分别为16 PFLOPS、2.9 PFLOPS 和5.9 PFLOPS ,相应于3.6%、1.5% 和 0.5%的峰值)。因此,DGDFT具有极快的计算速度,比目前最先进的高标度DFT-FE软件(采用23 K GPUs模拟11K原子)快1-2个量级;模拟尺度更是远超同类软件(如VASP),在保证化学精度的前提下可以模拟250万原子金属体系。
未来,随着超级计算机算力达到10E级,DGDFT软件的高可扩展性可以将模拟的体系进一步提升,可以把第一性原理材料模拟进一步扩展到宏观尺度(>1000nm),从而实现对真实材料和器件的模拟,为第一性原理材料模拟软硬件一体化的工业应用铺平道路。
瀚海量子

第一性原理(first-principles)电子结构计算的主要任务就是从量子力学基本原理出发,在无任何经验参数的条件下通过现代计算机从头计算分子、固体和纳米材料等的物理化学性质及其应用。该研究方法不需要开展真实的实验,极大地节省了实验成本,缩短了新材料的开发周期,为材料的制备和改性、新材料的开发以及极端环境下材料的性质研究提供了有效的理论指导。因此,第一性原理电子结构计算是二十一世纪解决实验理论问题和预测新材料结构性能的强有力工具和标准研究方法。
合肥瀚海量子专业从事基于量子力学的第一性原理电子结构计算和分子动力学模拟等软件研发,以中国科学技术大学杨金龙院士课题组胡伟研究团队负责运营推广的多款软件产品为核心产品,致力于打破国内计算软件市场被国外软件垄断的僵局。瀚海量子的软件采用了目前国际上最先进的低标度数值计算方法和迭代对角化算法,结合软件硬件一体化高性能并行计算设计,尤其是针对国产超级计算机的异构并行框架,优于本领域其它所有计算软件,是目前做第一性原理固体分子材料计算体系最大(最高250万原子)、效率最高(支持GPU-CUDA加速,并全面适配国产超算)的计算软件,可赋能新材料、新能源、生物制药、人工智能、量子力学科普科教等多个领域。


